Projects
Projects in Genetics
Decentralized GWAS
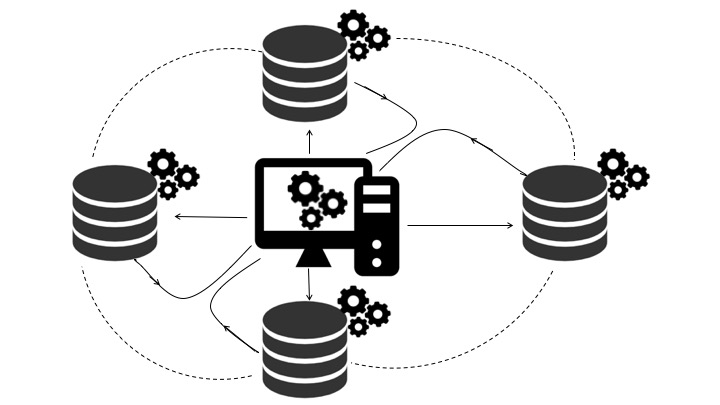
Genome Wide Association Studies GWAS aim to tease out associtations between single nucleotide polymorphism (SNP) (mutations of a single basepair) and a particular disease or phenotype. Because a phenotype is often influenced by many SNPs, often a large sample size is needed to achieve statistical power. The two common approaches are 1) to gather the required amount of data and analyze the data in one location, or 2) use meta-study techniques. The first approach leads to significant loss in privacy of participants and can have other associated costs. The second approach can lead to bias. In this project, we implemented a decentralized end-to-end GWAS pipeline with population structure control via PCA. The manuscript is currently under preparation. Recent presentation to Genome Sequencing Project. Paper
Dimensionality reduction in PopGen
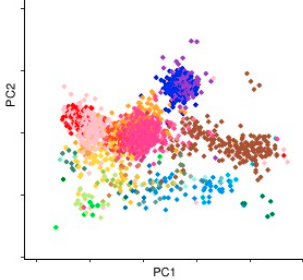
The picture to the left shows a PCA plot of the Population Reference Sample (POPRES) from Nelson, et. al. 2008 data. Dimensionality reduction algorithms have been a main staple population genetics. PCA is the most common and often most biologically meaningful approach for dimensionality reduction on the genotype matrix (see McVean, 2009). Flashpca and EIGENSOFT provide scalable software for performing PCA, however, they do not handle more sophisticated PCA approaches that are often useful to address biases in the data. For and example see McVean, 2009 for how lack fo balance in samples from each region can lead to bias. This project aims at producing a software package that interfaces with the common input data formats, addresses the aforementioned bias via, weighted PCA, debiases ancient DNA projections, provides other exploratory dimensionality reduction approaches and works with data collected on different arrays. As opposed to the two aforementioned softwares that aim at biobank size datasets, this package is aimed for use with small and medium size cohorts. The accompanied tutorial will be linked here. This project is currently in progress.
Clinical Genetics
In a clinical setting, a slew of sources of information are investigated in order to classify a mutation as pathogenic or benign (see Fig. 1 of Richards et. al.). The boundaries for scoring different sources is determined based on clinical intuition. Therefore, the bounderies for different sources of information may not be consistent. My goal is to combine a data driven approach (ordinal logistic regression) with this clinical intuition (determining the ordinal variables) to provide consistent boundaries for all sources evidence.
Networks in biology
Computational Denoising
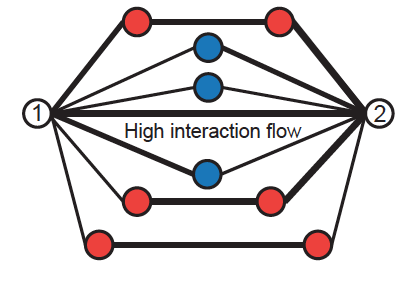
For many biological datasets, Networks provide a nice abstraction of the dynamics and relations between the pieces. Biological data is often noisy, due to both biological and technical noise. Reducing the noise, if possible, can be costly. On the other hand, working with noisy data can lead to inaccurate conclusions. One remedy is to use biological intuition to denoise the data. I've been involved with two projects in computational denoising of biological data. My main contribution is in developing Network Enhancement. (to appear in Nature Communications) we used a combination of diffusion and regularization for denoising generic biological networks. The intuition for diffusion is that in biology two elements being closed to the same element often implies that the initial two elements are close. On the other hand, regularization leads to a more sparse network and controls the scale. We show that mathematically, our algorithm leads to a "smoothed out" PCA, meaning that smaller singular values are shrunk more aggressively. I've also been involved in developing a Hi-C network denoising algorithm presented here.
Microbiome
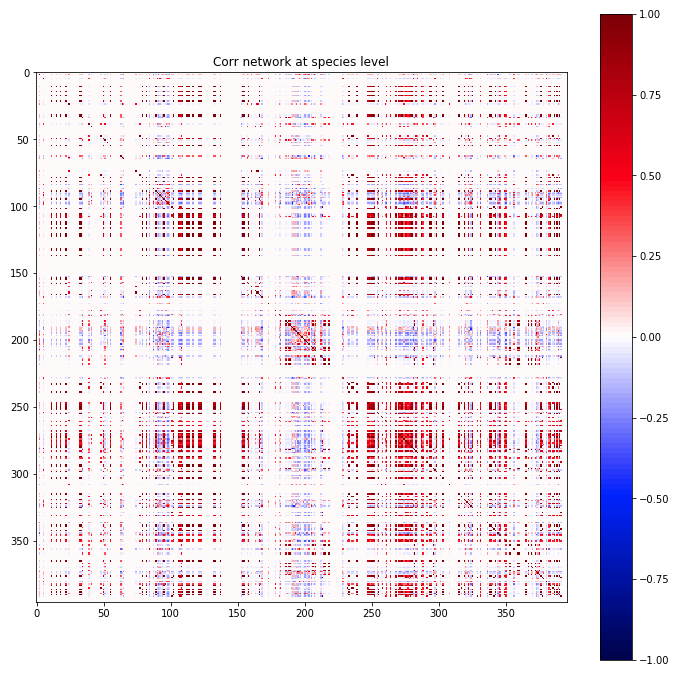
Understanding the gut microbiome is a promissing angle for understanding and treating obesity and digestive disease (see Thurnbaugh et. al. for example). In this path, understanding the interactions and correlations between microbiota can be an important first step. Unfortunately, the sheer number of participating microbes, the wide range in prevalence, and identificaiton problems leads to noisy measurements (as seen to the right). I am exploring regularization approaches that borrow information from time-series data, other participants, and phylogenetic tree that smooth out the noise and lead to more generalizable conclusions. As part of a collaboration, we are also studying the network properties of microbiome data (centrality, motif counts, path lengths, etc.). This project is still in the exploration phase.
